Research fields
Our research group is dedicated to cutting-edge research in the field of computational chemistry.
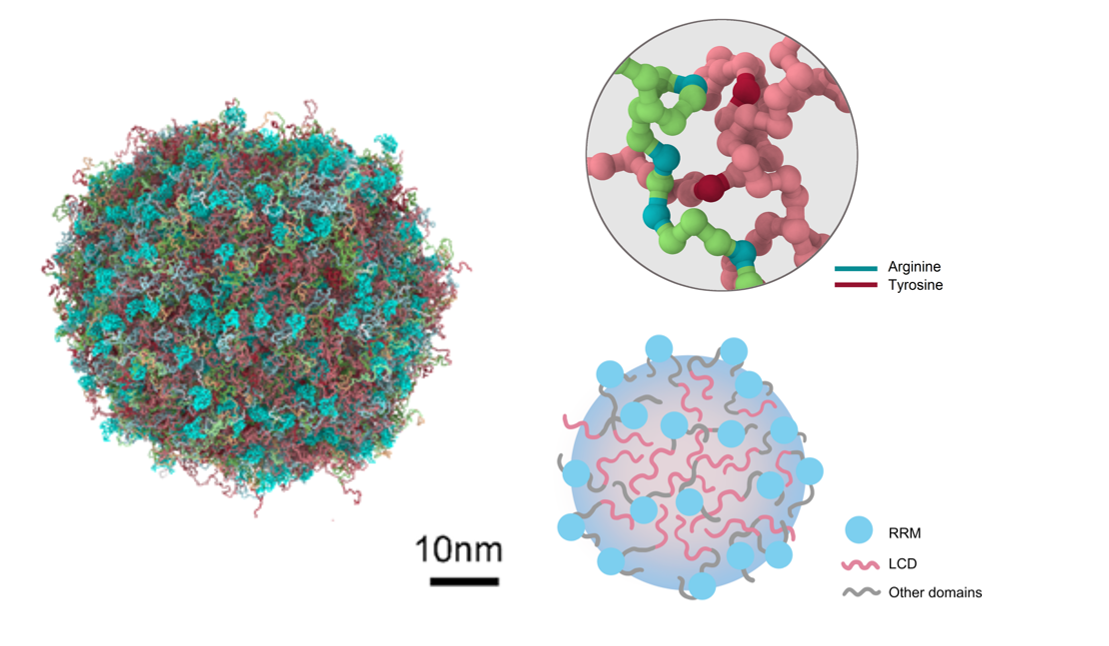
Simulation Study on Biomolecular Condensates
IDRs, RNAs and other macromolecules undergo liquid–liquid phase separation driven by multivalent, weak interactions, instantly erecting membrane-less organelles that orchestrate myriad cellular processes. Using large-scale coarse-grained molecular dynamics, we resolve nucleation, growth and network maturation in atomistic detail, mapping sequence-encoded phase diagrams, interfacial curvatures and dynamic topologies to uncover how molecular grammar, valency patterning and post-translational edits jointly dictate condensate architecture, material state and emergent function. For further information please contact Prof. Li Zhao.
Biology
CGMD